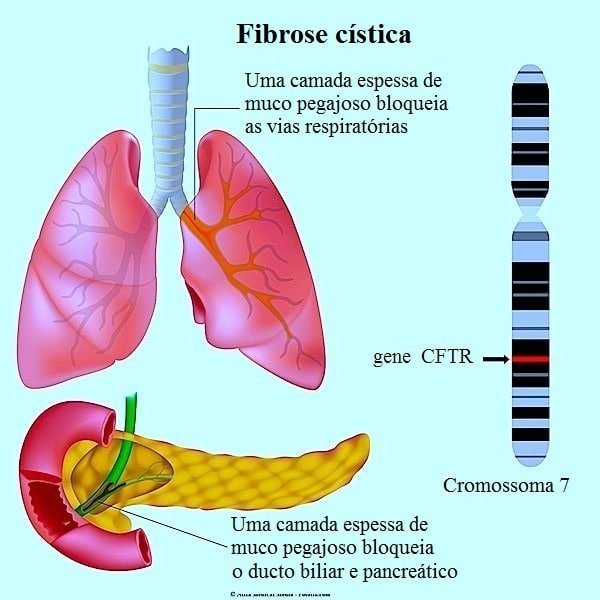
A fibrose cística (FC), ou mucoviscidose, é uma doença geneticamente determinada decorrente de comprometimento da função da proteína CFTR (de cystic fibrosis transmembrane regulator), envolvida com o transporte transmembrana de cloro e bicarbonato.
O gene CFTR foi identificado em 1989 e mais de 1.000 diferentes mutações que ocasionam alteração em seu funcionamento já foram até o presente identificadas
. Uma delas, conhecida como delta F508 (por levar a deleção do aminoácido fenilalanina na posição 508), é encontrada em 70% dos cromossomos dos caucasóides com FC.
Algumas outras mutações são encontradas de forma recorrente em determinadas populações, mas muitas delas são ditas privadas, ou seja, observadas até o momento em uma única família.
Principais características fenotípicas:
- Idade de início: neonatal à vida adulta
- Doença pulmonar progressiva
- Insuficiência pancreática exócrina
- Azospermia obstrutiva (ausência bilateral de ductos deferentes)
- Concentração elevada de cloreto no suor
- Íleo meconial (obstrução do íleo terminal por mecônio anormalmente endurecido)
- Doença predominante dos norte-europeus
Traços mais comuns na história e exame físico:
- Déficit de crescimento (resulta de uma combinação de consumo aumentado de calorias devido a infecções pulmonares crônicas e subnutrição devido à insuficiência pancreática exócrina)
- Presença de cólica e diarreia
- Síndrome da má absorção
- Tosse crônica
- Repetidas infecções do trato respiratório superior
Fisiopatologia
Distúrbio autossômico recessivo do transporte iônico epitelial causado por mutações no gene CFTR, que é um canal iônico que conduz cloreto e bicarbonato. O CFTR facilita a manutenção da hidratação das secreções das vias aéreas, através do transporte de cloreto e inibição da captação de sódio.
As secreções desidratadas e viscosas nos pulmões dos pacientes com FC interferem com a depuração mucociliar, inibem a função dos peptídeos antimicrobianos que ocorrem naturalmente, proporcionam um meio de crescimento para os organismos patogênicos e obstruem o fluxo de ar.
A perda do transporte de cloreto pelo CFTR no ducto pancreático prejudica a hidratação de secreções e leva à retenção de enzimas exócrinas no pâncreas. O dano provocado por essas enzimas retidas causa, por fim, a fibrose do pâncreas.
Diagnóstico
Normalmente baseia-se em critérios clínicos e na concentração de cloreto no suor.
Canal norteamericano com muitas informações sobre fibrose cística:
Segue as Notícias da Comunidade PortalEnf e fica atualizado.(clica aqui)