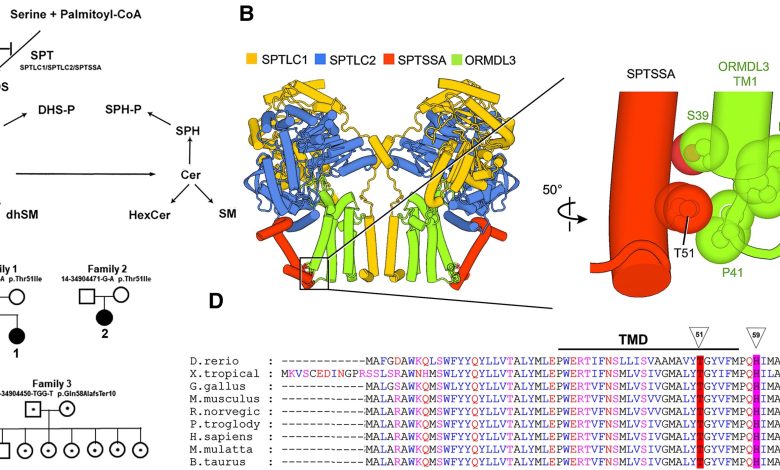
 A SPT catalisa a primeira etapa da síntese de esfingolipídios, a condensação de serina com um acil-CoA (normalmente, palmitoil-CoA) para gerar a base esfingoide, 3-KDS, que é posteriormente modificada para formar o complexo família de SLs. SPT é feedback inibido pelas proteínas ORMDL. Os SLs à base de diidroceramida [dihydroceramide (dhCer), dihydrohexosylceramide (dhHexCer) and dihydrosphingomyelin (dhSM)] geralmente não se acumulam em níveis apreciáveis, mas a atividade excessiva do SPT resulta em elevação significativa desses SLs. (B) O complexo SPT/ORMDL3 é um dímero de tetrâmeros SPTLC1/SPTLC2/SPTSSA/ORMDL3. A imagem ampliada (à direita) mostra que Thr51 de SPTSSA está em contato próximo com a extremidade luminal do primeiro (de quatro) domínio transmembranar (TM1) de ORMDL3. (C) Pedigrees mostrando a ocorrência de novo da variante heterozigótica SPTSSA p.Thr51Ile em duas famílias e a variante herdada p.Gln58AlafsTer10 na terceira família. Os símbolos preenchidos denotam probandos; pontos no centro do círculo ou quadrado denotam portadores. (D) Thr51 (indicado pelo triângulo) da subunidade SPTSSA evolutivamente conservada está localizada na extremidade luminal do domínio transmembranar único (TMD).")
Biossíntese de SL e variantes causadoras de HSP na subunidade SPTSSA de SPT. (A) A SPT catalisa a primeira etapa da síntese de esfingolipídios, a condensação de serina com um acil-CoA (normalmente, palmitoil-CoA) para gerar a base esfingoide, 3-KDS, que é posteriormente modificada para formar o complexo família de SLs. SPT é feedback inibido pelas proteínas ORMDL. Os SLs à base de diidroceramida [dihydroceramide (dhCer), dihydrohexosylceramide (dhHexCer) and dihydrosphingomyelin (dhSM)] geralmente não se acumulam em níveis apreciáveis, mas a atividade excessiva do SPT resulta em elevação significativa desses SLs. (B) O complexo SPT/ORMDL3 é um dímero de tetrâmeros SPTLC1/SPTLC2/SPTSSA/ORMDL3. A imagem ampliada (à direita) mostra que Thr51 de SPTSSA está em contato próximo com a extremidade luminal do primeiro (de quatro) domínio transmembranar (TM1) de ORMDL3. (C) Pedigrees mostrando a ocorrência de novo da variante heterozigótica SPTSSA p.Thr51Ile em duas famílias e a variante herdada p.Gln58AlafsTer10 na terceira família. Os símbolos preenchidos denotam probandos; pontos no centro do círculo ou quadrado denotam portadores. (D) Thr51 (indicado pelo triângulo) da subunidade SPTSSA evolutivamente conservada está localizada na extremidade luminal do domínio transmembranar único (TMD).
Uma mutação genética no gene SPTSSA é identificada como a causa da paraplegia espástica hereditária, uma doença rara que causa fraqueza progressiva, rigidez e espasticidade nas extremidades inferiores, de acordo com um estudo publicado no Cérebro. O gene SPTSSA é responsável por estimular a serina palmitoiltransferase, ou SPT, uma enzima com funções críticas no sistema nervoso.
Os pesquisadores da Uniformed Services University of the Health Sciences (USU), em colaboração com o Massachusetts General Hospital (MGH), descobriram que dois dos três pacientes não aparentados com um caso complexo de HSP tinham a mesma mutação no gene SPTSSA. Em ambos os casos, esta foi uma nova mutação que seus pais não tiveram. Essa mutação altera uma única unidade na cadeia de aminoácidos, que compõem a proteína SPTSSA.

No terceiro paciente, um jovem com um caso menos grave de HSP, os pesquisadores descobriram uma mutação herdada no SPTSSA, fazendo com que sua proteína SPTSSA fosse mais curta do que o normal. Seus pais assintomáticos e vários irmãos não afetados tinham uma cópia mutante e uma cópia normal do gene SPTSSA, e esse paciente herdou a cópia mutante do gene SPTSSA de cada pai.
Os pesquisadores determinaram que essas variantes SPTSSA causadoras de doenças impedem a regulação normal da síntese de esfingolipídios. SPTSSA é uma subunidade de uma enzima – SPT ou serina palmitoiltransferase. A enzima SPT catalisa o primeiro passo na produção de esfingolipídios – uma família diversificada de lipídios com funções celulares críticas que são altamente enriquecidas no sistema nervoso.
Mutações em outras subunidades do SPT são conhecidas por causar outros doenças neurológicas, como HSAN1, o tipo mais comum de neuropatia periférica hereditária e uma forma juvenil de ALS. Se a regulação do SPT estiver funcionando adequadamente, quando os níveis de esfingolipídeos se tornam muito altos, eles se ligam ao complexo SPT/ORMDL e inibem a atividade do SPT. No entanto, as variantes do SPTSSA prejudicam essa regulação e levam a uma atividade irrestrita do SPT. Isso resulta em uma superprodução de esfingolipídios que leva a doenças neurodegenerativas.
Embora apenas um pequeno número de pacientes seja afetado – cerca de 20.000 pessoas nos EUA sofrem de HSP – o impacto nas famílias é enorme. Além da espasticidade progressiva dos membros, que caracteriza o HSP “puro” – onde os músculos enrijecem ou apertam devido a uma ruptura entre o cérebro e a medula espinhal, os pacientes com uma forma complexa da doença, incluindo esses pacientes com SPTSSA, também exibem alterações sensoriais e envolvimento cognitivo.
Até agora, mais de 80 mutações genéticas têm sido associados a várias formas de HSP, mas este é o primeiro relato de que a homeostase esfingolipídica perturbada causa essa doença.
Essas descobertas reforçam o crescente corpo de evidências de que mutações nas subunidades do SPT podem levar a diferentes tipos de doenças neurológicas. Eles também deixam claro que os distúrbios na síntese de esfingolipídeos estão por trás de várias doenças neurológicas e que a correção da homeostase dos esfingolipídeos pode ser usada como uma estratégia de tratamento potencial para essas doenças devastadoras.
“Estamos extremamente empolgados com essas descobertas, que são consistentes com nossos estudos anteriores sobre variantes causadoras de ELA juvenil”, disse a Dra. Teresa Dunn, co-autora principal do estudo e presidente do Departamento de Bioquímica e Biologia Molecular da USU.
“Juntos, este estudo e nossas pesquisas anteriores serão extremamente importantes para entender como a regulação perturbada dos níveis de esfingolipídios leva a doenças neurodegenerativas e levanta a possibilidade de que outros genes associados com HSP e ALS desempenhem um papel na manutenção da homeostase dos esfingolipídeos. Esses estudos também apontam para esfingolipídios como potenciais biomarcadores e sugerem possíveis estratégias de tratamento”.
Este estudo também é importante para fornecer pistas sobre quais vias metabólicas estão envolvidas em muitas outras doenças com sintomas clínicos semelhantes, acrescentou Dunn.
O SPTSSA foi descoberto pela primeira vez por Dunn e colegas de trabalho há uma década. Estudos em andamento no laboratório de Dunn mostraram que o SPTSSA desempenha um papel na inibição por feedback do SPT por proteínas chamadas ORMDLs que regulam a função do SPT. Como resultado, quando as variantes do SPTSSA foram descobertas em pacientes, a equipe estava bem posicionada para determinar o mecanismo da doença, explicou Dunn.
“Esta descoberta desses defeitos genéticos encerra a odisseia diagnóstica para várias famílias afetadas por problemas motores progressivos. Reconhecer a importância da regulação dos esfingolipídios durante o desenvolvimento inicial do cérebro inaugura um novo capítulo na neurobiologia”, disse o Dr. Florian Eichler, neurologista e diretor do Serviço de Leucodistrofia do MGH.
O estudo foi liderado por Dunn da USU e pelo Dr. Florian Eichler do Hospital Geral de Massachusetts, um neurologista pediátrico, em colaboração com a Undiagnosed Disease Network (UDN) e várias outras universidades, incluindo Duke University, Baylor University e Hebrew University.
Mais Informações:
Siddharth Srivastava et al, as variantes do SPTSSA alteram a síntese de esfingolipídios e causam uma paraplegia espástica hereditária complexa, Cérebro (2023). DOI: 10.1093/brain/awac460
Fornecido pela Uniformed Services University
Citação: Mutação genética ligada à paraplegia espástica hereditária (2023, 31 de janeiro) recuperada em 1º de fevereiro de 2023 em https://medicalxpress.com/news/2023-01-genetic-mutation-linked-hereditary-spastic.html
Este documento está sujeito a direitos autorais. Além de qualquer negociação justa para fins de estudo ou pesquisa privada, nenhuma parte pode ser reproduzida sem a permissão por escrito. O conteúdo é fornecido apenas para fins informativos.
Segue as Notícias da Comunidade PortalEnf e fica atualizado.(clica aqui)